The Chemistry of Nitrogen Compounds
Dr David A.Widdowson
Course Content
1. Introduction
1.1 Scope
1.2 Coverage
1.3 Course Objectives2. Nitro Alkanes and Nitro Arenes
2.1 Structure
2.2 Electronic Effects
2.3 Spectroscopy
2.4 Synthesis of Nitro Compounds2.4.1 Gas Phase Nitration of Alkanes2.5 Reactions
2.4.2 Electrophilic Nitration of Anions
2.4.3 Nitrate SN2 Displacement of Alkyl Halides
2.4.4 Oxidation with Peracids
2.4.5 Aromatic Nitro Compounds2.5.1 a Anions2.6 Summary
2.5.2 Alkylation2.5.2.1 Henry Reaction2.5.3 Reduction
2.5.2.2 Michael Additions
2.5.2.3 Ambident Nucleophiles2.5.3.1 General
2.5.3.2 Photochemically
2.5.3.3 Metal/H+ Reaction
2.5.3.4 H2/Pd Surface Reactions
2.5.3.5 Sulphur Reagents
2.5.3.6 Metal Hydrides3. Nitroso Compounds
3.1 Structure
3.2 Electronic Effects
3.3 Spectroscopy
3.4 Synthesis3.4.1 Aromatics
3.4.2 Aliphatics
3.4.3 Tertiary Aliphatics3.5 Reactions3.5.1 Addition and Condensation
3.5.2 Reduction
3.5.3 Oxidation
3.5.4 Radical Addition-Spin Trapping3.6 Summary4. Amines
4.1 Nomenclature
4.2 Structure
4.3 Electronic Effects
4.4 Spectroscopy
4.5 Physiological Effects
4.6 Synthesis4.6.1 Alkylation Reactions3.6.1.1 General4.6.2 Reductive Methods
3.6.1.2 Alkylation of Phthalimide Anion-The Gabriel Synthesis
3.6.1.3 Alkylation of Bis(Phenylthio)Amine Anion
3.6.1.4 Alkylation of Trifluoromethanesulfonylamine3.6.2.1 Reduction of Amides4.6.3 Hydrolytic Methods
3.6.2.2 Reduction of Nitro Compounds or Azides
3.6.2.3 Reduction of CN Multiple Bonded Systems
3.6.2.4 Eschweiler-Clarke Synthesis3.6.3.1 Hydrolysis of Amides4.6.4 Rearrangements
3.6.3.2 MeOH/BF3.Et2O
3.6.3.3 Meerwein's Salt3.6.4.1 General Reaction Scheme
3.6.4.2 Hoffmann Rearrangement
3.6.4.3 Curtis Rearrangement
3.6.4.4 Lössen Rearrangement
3.6.4.5 Schmidt Reaction
3.6.4.6 Beckmann Rearrangement
3.6.4.7 Abnormal Beckmann Rearrangement4.7 Reactions4.7.1 Basicity and Nucleophilicity
4.7.2 Alkylation
4.7.3 Acylation
4.7.4 Halogenation4.7.4.1 Aliphatics
4.7.4.2 Aromatics4.7.5 Nitrosation4.7.5.1 General
4.7.5.2 Primary Amines
4.7.5.3 Secondary Amines
4.7.5.4 Tertiary Amines4.7.6 Reaction with Carbonyls
4.7.7 Oxidation4.7.7.1 Oxygen Addition Reactions
4.7.7.2 Dehydrogenation of Amines4.7.8 Deprotonation4.8 Summary5. a Amino Acids, Peptides and Proteins
5.1 Amino Acid Nomenclature
5.2 Amino Acid Structure
5.3 Amino Acid Synthesis5.3.1 The Gabriel Synthesis
5.3.2 The Streaker Synthesis
5.3.3 Enantioselective Synthesis5.4 Peptide and Protein Structure
5.5 Peptide Synthesis5.5.1 General
5.5.2 Merrifield Solid Phase Synthesis
5.5.3 Peptide Synthesis in Nature5.6 Summary
6. Nitrogenous Natural Products and Alkaloid Biosynthesis
6.1 Introduction
6.1.1 Amino-acids, Peptides, Proteins and Derived Substances
6.1.2 Vitamins
6.1.3 Porphyrins, Chlorins and Cobalamins
6.1.4 Purines, Pyrimidines, Nucleosides, Nucleotides6.2 Alkaloid Biosynthesis
Introduction
Scope
There are many organic functional group which contain nitrogen, the scope of this course will be;- Nitro compounds, RNO2
- Nitroso compounds, RNO
- Amines R3N and their derivatives
- Amino acids, peptides and proteins
- Other nitrogenous natural products - alkaloids and nucleotides
Coverage
For each of these the following area will be considered;- Structure and stereochemistry
- Physical data and spectroscopic characteristics
- Synthesis
- Chemical reactivities and relationship to electronic properties
Course Objectives
To compare the overview of the common functional groups of organic chemistry by;- Extending the first year functional group chemistry to N-compounds.
- Explaining the effect of the heteroatom/group on the structure of organic molecules.
- Discussing the effect of the heteroatom/group on the reactivity of organic molecules.
- Rationalising the effect of the heteroatom/group on the transformations of organic molecules.
- Relating the nitrogen chemistry to polyfunctional molecules through an introduction to nitrogenous natural products
Nitro Alkanes and Nitro Arenes
Structure
The nitrogen is trigonal planar with a bond angles of 120°, there are two resonance forms so implying that the two oxygens are equivalent;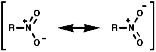
Electronic Effects
These are strongly electron withdrawing both inductively, -I, and mesomerically, by resonance, -R. This means that both the C-N s bond and the p system are strongly polarised, Cd+-NO2d-.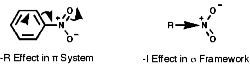
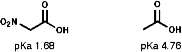
Spectroscopy
I.R.The nmax for the N=O stretch is 1500-1600 cm-1, compared to the C=O stretch at 1650-1800 cm-1. NMR
For a CH protons, adjacent to the group, the chemical shift, d, = 4.3, due to electron withdrawing effect;
The nitro group causes a pronounced shift of lmax to longer wavelengths when conjugated to unsaturated p systems, a bathochromic shift. This is why nitro compounds are often yellow.
Synthesis of Nitro-Compounds
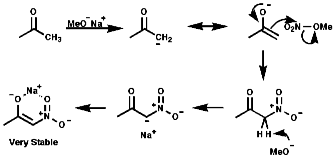
Aliphatic nitro compounds are synthesised by; Gas Phase Nitration of AlkanesThis commercial free radical process involves the NO2 radical;
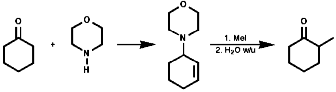
The use of the enolate active methylenes forms a stable product due to the chelation of the counter ion;
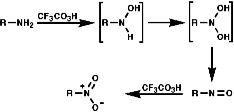
Oxidation with Peracids
Nitro compounds can be produced by oxidised of amine by peracids;
These are synthesised by electrophilic aromatic substitution with NO2+ ions;
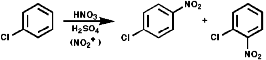
Reactions
As the nitro group is strongly electron withdrawing and shows affinity with the C=O group. Thus addition across the N=O is possible and reduction is easy. a Anionsa anions are easily formed with base and stabilised by resonance as nitronate anions, c.f. C=O enolate formation;
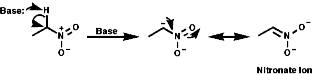
As the nitronate ion is delocalised it is a soft nucleophile and show usual reactivities of stabilised, soft, carbanions.
Alkylation
This reaction is analogous to the Aldol reaction;
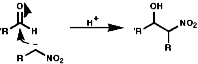

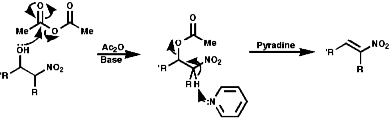
The Michael addition is a conjugate addition as the double bond is the soft centre of the ester, the carbonyl carbon being the hard centre. It proceeds by the following mechanism;
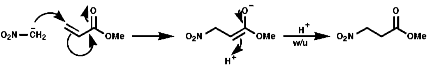
Nitronate anions themselves can act as ambident nucleophiles with either attack from the C, a soft nucleophile, or from the O, hard nucleophile. These will attack soft or hard electrophiles respectively;
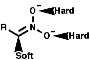
Reduction
General
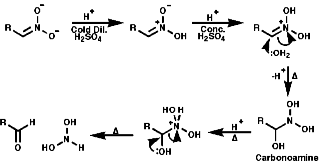
In principle the reduction of nitro compounds should follow the path;
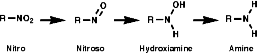
Photochemically
Metals, such as Fe, Zn, Sn can be used with H+ to reduce the nitro group by a sequence of single electron transfer (SET)/protonation reactions;
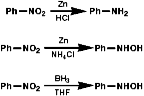
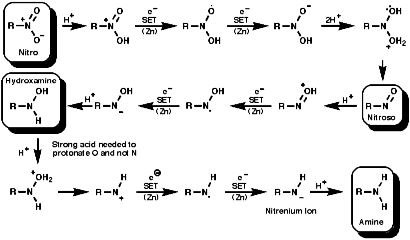
Reactions H2/Pd or Pt can be used by heterogeneous H- transfer on the metal surface and then reaction with the nitro compound;
NaSH or Na2Sx or (NH4)2S2 can be used and possibly precede by SET reactions;
Reagents like LiAIH4, but not usually NaBH4, reduce the nitro group by hydride, H-, transfer. The product will depend on the nature of the reducing agent, but the end point is ultimately an amine.
Summary
- N SP2 hybridised and strongly electron withdrawing
- H acidic
- Anion reacts with E+, RBr RCHO, Henry reaction etc.
- Soft anion soft undergoes Michael additions
- Group reduced by:
- Metal/H+ (electron source)
- H-, (NaBH4 or LiAlH4)
- H2/Pd or Ni (R)
Nitroso Compounds
Structure
The nitrogen is trigonal planar with a bond angle of ª125°. Nitroso compounds tend to dimerise in the following way;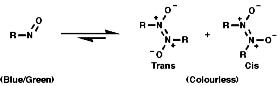
Electronic Effects
Nitroso groups are strongly electron withdrawing, like the nitro group, but the situation is complicated by the dimerisation reaction.Spectroscopy
I.R.The nmax for the N=O stretch in the monomer is 1560 cm-1, in the dimer the nmax for the N-O stretch is 1200 cm-1. NMR
For a CH protons, adjacent to the group, the chemical shift, d, = 4.0, due to electron withdrawing effect.
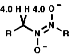
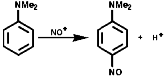
Synthesis
AromaticsThe synthesis of aromatic nitroso compounds can achieved by nitrosation with NO+, form HNO2, on strongly activated, electron rich, aromatics like ArNH2 and ArOH;
Aliphatics
The synthesis of aliphatic nitroso compounds can achieved by nitrosation of active, acidic, methylene compounds under acidic conditions, H+/NOCl, N2O3 or N2O4;
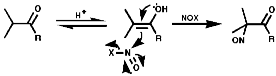
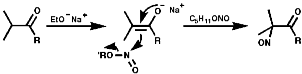
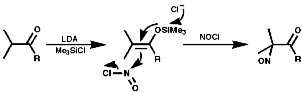
Tertiary Aliphatics
To synthesis nitroso compounds with tertiary alkyls another method has to be employed;
Reactions
Addition and CondensationAs the nitrosyl group is strongly electron withdrawing and more similar to the C=O than the NO2 group There is polarisation of the N=O bond and so behaves as a weak C=O. It undergoes addition of nucleophiles and condensation with primary amines and the anions of active methylene compounds, e.g malonates, b ketoesters;
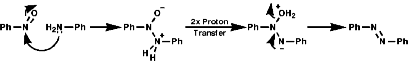
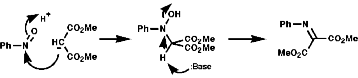
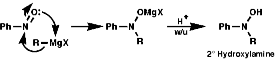
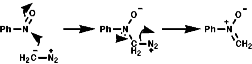
Reduction occurs as for NO2 groups with metals, metal hydrides, hydrogen/ catalyst.
Oxidation is readily brought about with peracids inter alia, this is not possible for nitro groups.
Monomeric nitroso compounds are used to detect transient free radicals, mainly carbon centred radicals, by 'trapping' them as stable nitroxide radicals. These can be detected and assayed in an EPR spectrometer, this is the electron equivalent of the NMR spectrometer. EPR stands for Electron Paramagnetic Resonance. This allows the intermediacy of such otherwise undetectable transients to be proven and is therefore a valuable mechanistic probe.
If the following thermal decomposition of benzoyl peroxide, a radical initiation reaction, is to be monitored;
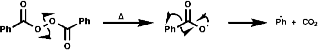
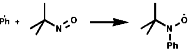
Summary
- N SP2 hybridised and electron withdrawing
- ON group like carbonyls undergoes addition with RMgX, condenses with RNH2 and 'active methllene', e.g malonate anion
- When a-H present, readily isomerises to oxime
- Very rapid addition of radicals R across N=O bond to give stable nitroxide radicals
Amines
Nomenclature
- R-NH2 Primary Amide
- R2-NH Secondary Amide
- R3-N Tertiary Amide
- R4-N+ Quaternary Ammonium Ion
Structure
The Nitrogen is SP3 hybridised but rapidly inverting, for NH3 the inversion rate is = 2 x 1011;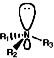
Electronic Effects
The nitrogen in an amine is both electronegative and able to donate its loan pair into a p system. Therefore it has both -I and +R effects, the +R effect is greater than the -I effect though.Spectroscopy
I.R.The nmax for the N-H stretch as 3300-3400 cm-1. NMR
For protons in the a position to a primary amine, e.g -CHNH2, the protons have a chemical shift of d 2.5-3. Protons in the a position to a tertiary amine, e.g CH3NR2, have a chemical shift of d 2.0-2.5. Protons in the a position to a quaternary ammonium ion, e.g -CHN+R3, have a chemical shift of d 3.5 - 4.0.
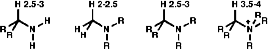
Physiological Effects
Amines are an important group of compound intimately concerned in many biological processes, not only at the a amino acid protein level. Because of their hydrogen bonding properties, amines are much involved in the binding polar molecules to macromolecules as in enzyme-substrate and hormone-receptor interactions. Consequently, very many medicines and crop protection agents, which frequently act as antagonists or agonists for natural substances, contain basic nitrogen. Similarly the alkaloids, which are by definition natural nitrogenous compound, are much involved in folk medicine and in natural biocides.Synthesis
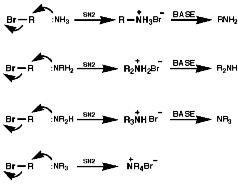
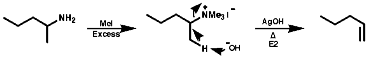
Because of the widespread importance of this functional group, many methods of preparation of the various types of amine have been devised. These can be classified as; Alkylation ReactionsThe synthesis of primary, secondary, tertiary and quaternary amines involving direct alkylation of ammonia and lower amines. The reaction proceeds with nucleophilic attack by the loan pair on nitrogen and then abstraction of the extra proton with a base that is more nucleophilic than the amine. It is not generally an efficient process because of ready over reaction;
Alkylation of Phthalimide Anion-The Gabriel Synthesis
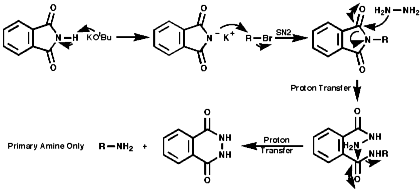
Hydrazionlysis of the product releases the primary amine.
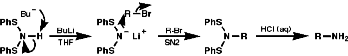
Acid cleavage again gives the primary amine.
Cleavage with LiAlH4, or base, gives the secondary amine.
- Reduction of Amides
This can be achieved with LiAlH4 or better still the electrophilic BH3.THF.
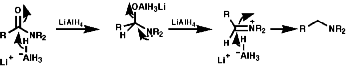
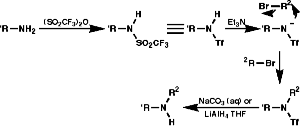
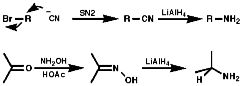
- Reduction of Nitro Compounds or Azides
Reduction can be achieved with LiAlH4 or H2/Pd. The aliphatic substances can be synthesised by nucleophilic displacement of X from RX with NO2-R or N3-R.
- Reduction of CN Multiple Bonded Systems
The reduction of compounds such as, RC=N, R2C=NOH, can be achieved with LiAlH4 or H2/Pd.
- Eschweiler Clarke Synthesis
This involves the reductive alkylation of in situ generated RN=CR2 with HCO2- or NaBH3CN;
This reaction requires acidic conditions to produce the protonated form of the first product. This means that a mild hydride, H-, source is required an example of this is HCO2H/HCO2-Na+;
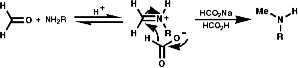
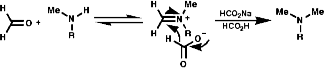
It is also possible to use NaBH3CN which is acid stable at pH 4.Hydrolytic Methods
- Hydrolysis of Amides
- MeOH/BF3.Et2O
- Meerwein's Salt
Simple hydrolysis of the amide with H3O+ or OH- is possible;
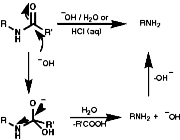
If R is aryl then it may be quite resistant to hydrolysis, a method of mild non-aqueous acid hydrolysis can then be used;
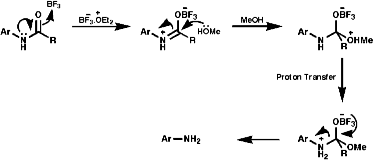
Meerwein's salt, Et3O=BF4-/H2, is used for mild hydrolysis of acid sensitive amides such as as b lactam side chains, this is an important reaction for the synthesis of penicillins;
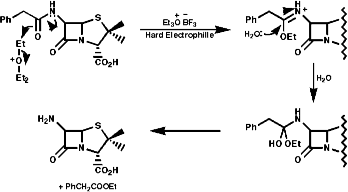
- General Reaction Scheme
- Hoffmann Rearrangement,Y=Br
- Curtius Rearrangement,Y=N2
- Lössen Rearrangement,Y=OAc
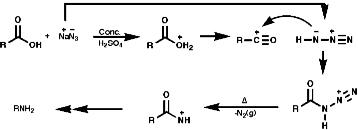
- Schmidt Reaction,Y=N2
- Beckmann Rearrangement
- Abnormal Beckman Rearrangements
A group of similar reactions involve the migration of an alkyl group, R, from carbon to electron deficient nitrogen, the nitrene or nitrenium ion. The nitrene is produced from the loss of a good leaving group, Y. The products, isocyanates, are readily hydrolysed to amines;
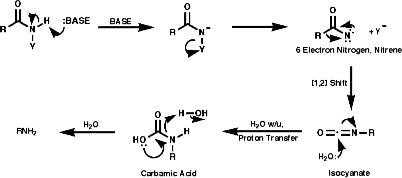
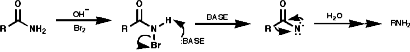
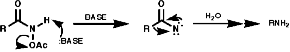
This is a rearrangement of ketoximes, RC(=NOH)R';
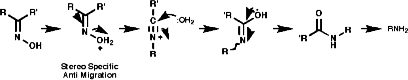
This occurs with the oximes of aldehydes and other carbonyls in which R or R' in R (=NOH)R' can form stable carbocations;
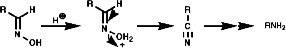
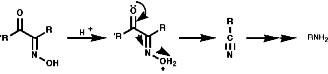
Reactions
Basicity and NucleophilicityThe lone pair of electrons on the nitrogen atom renders the amine basic and nucleophilic. The pKa's of the conjugate acids of simple amines increase with increasing alkyl substitution, this is due to the electron release from the alkyl groups, up to the di-alkylamine level. Thereafter the pKa diminishes because of increasing steric hindrance to protonation. The larger the pKa the stronger the base. Some pKa's of some conjugate acids in water at 25°C are;
Amine |
pKa |
| NH3 |
|
| EtNH2 |
|
| Et2NH |
|
| Et3N |
|
| PhNH2 |
|
| PhNEt2 |
|
Alkylation
This has been largely discussed under synthesis. One important area is the alkylation of aryl amines which N-alkylate;
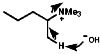
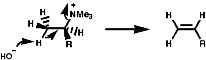
Acylation of primary and secondary amines with RCOX where X is a good leaving group like a halogen, OCOR' or OR group. It is a very common process, occurring via the tetrahedral addition product of the active carbonyl group;
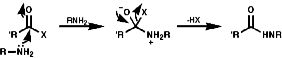
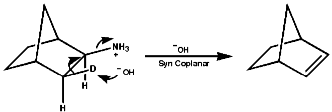
Aliphatics
N-Halo,Cl, Br, I, compounds are readily formed with Hal+ reagents;
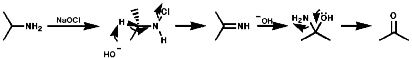
In benzene rings carbon attack of Hal+ is very common and has been dealt with before in the course. The amino group is electron releasing and o, p directing;
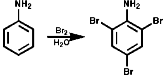
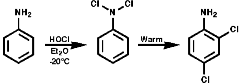
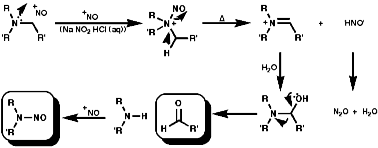
Nitrosation, with the active agent of NO+ from HNO, NOCl, N2O3, N2O4, is not subject to any significant steric inhibition, due to the small size of the NO+ group, and any nitrogen with a nucleophilic lone pair of electrons will react. The products depend on the nature of the amino function.
- Primary Amines
These react rapidly to generate diazonium ions R-N+=N. In the aryl series, ArN2+, these are stabilised by resonance with the aromatic ring. They have a relatively long lifetime and are often stable at room temperature;

They undergo nucleophilic attack by secondary amines at the terminal nitrogen to give the carcinogenic triazenes ArN=NNR2. Many other nucleophiles will react analogously;
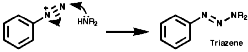
Fragmentation of the aryl diazonium ions can occur by ionic or single electron transfer, SET, radical mechanisms dependent upon the conditions. The copper (I) catalysed reactions, Sandmeyer reactions etc, proceed by the SET mechanism which generates the aryl radicals;
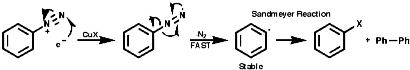
Thermal decomposition or reactions with simple nucleophiles are more likely to proceed via the ionic fragmentation to the highly reactive aryl cations;
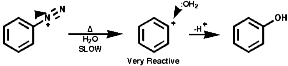
In either case, the overall product is the replacement of the N2+ group with a nucleophile, Hal-, CN-, OH- etc. Aliphatic diazonium ions are not stabilised and undergo very rapid fragmentation to N2 and an alkyl cation;
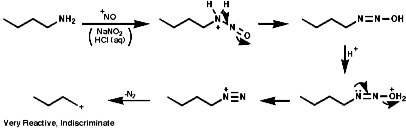
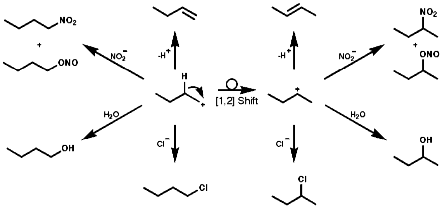
This species is itself very reactive and therefore indiscriminate in is reactions with nucleophiles. A plethora of products, including those resulting from rearrangement of the initially formed carbocation, are usually observed;
- Secondary amines
- Tertiary Amines
These readily form stable N-nitroamines, R2NN=O. N-Nitrosamines are potent carcinogens and their synthesis should only be undertaken under strictly controlled conditions;
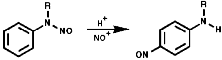
Aromatic tertiary amines, ArNR2, give directly the 4-nitroso-product;
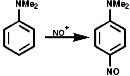
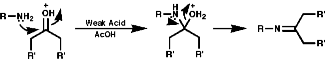
Primary amines simply condense to form imines;
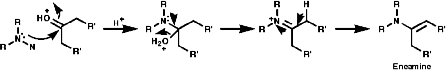
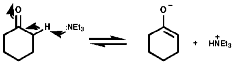
- Oxygen addition reactions
- Dehydrogenation of amines
These have been mentioned previously. Primary amines are oxidised by peracids, like trifluoroperacetic acid and perboric acid, to the nitro compounds;
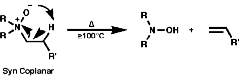
Tertiary amines in particular can be clearly dehydrogenated in the a position by oxidation with mercury salts like Hg(OAc2) or Hg(OCOCF3)2;
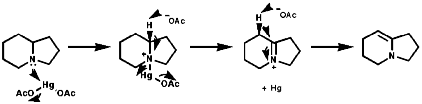
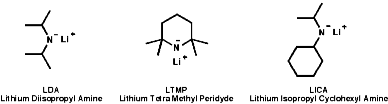
Primary and secondary amines can be deprotonated by very strong bases;
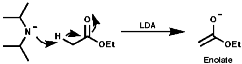
Summary
- N SP3, tetrahedral, normally rapidly inverting therefore achiral
- Electronically has -I (mainly s bond), +R ( p bond ) effects
- All reactivity, in this course, stems from the donation of electrons from the lone pair
- Synthesis by;
- alkylation of lower homologues
- Reduction of RCONR'2, RN3, RCN, RNO2, RCH+NOH etc
- Acyl nitrene rearrangements, Hoffmann, Lössen, Schmidt, Curtius
- Beckmann rearrangements
- Lone pair nucleophilic, reacts with many electrophiles, R-X, RCHO, R2CO. RCOX, Hal2, NO+,
- Lone pair basic, R2NH > R3N > RNH2 > NH3
a Amino Acids, Peptides and Proteins
Amino Acid Nomenclature
Although several hundred a amino acids have been detected in nature, only 21 are found in proteins. The others serve other purposes. Also, despite being less widespread, the D-series amino acids occur quite commonly, e.g in bacterial cell walls. They all have trivial names for a complete list see Vollhardt, 2nd edition. p.1025.
R |
Name |
R |
Name |
|
| -Me |
|
-CHMe3 |
|
|
| -CH2CHMe2 |
|
-CHMeCH2Me |
|
|
| -CH2Ph |
|
-CH2C6H4OH |
|
|
| -CH2OH |
|
-CHMeOH |
|
|
| -CH2CO2H |
|
-CH2CONH2 |
|
|
| -CH2CH2CO2H |
|
- CH2CH2CONH2 |
|
Amino Acid Structure
a amino acids are the basic building blocks of peptides, including peptide hormones, and proteins. They can be represented in many ways but the common ways are shown below;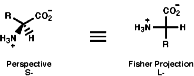

Amino Acid Synthesis
Many methods have been developed to synthesise amino acids, classically; The Gabriel SynthesisThe can be readily adapted for amino acids;
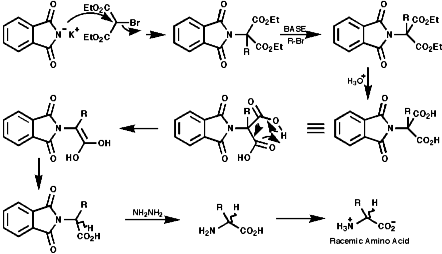
This is a direct approach, but not enantioselective;
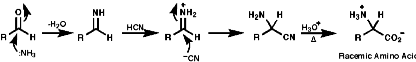
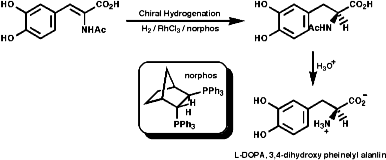
This modern method of making only one stereo isomer include the asymmetric hydrogenation of dehydroamino acid derivatives. For example the Monsanto synthesis of L-DOPA;
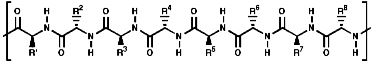
These are oligomers, peptides, and polymers, proteins, of the a amino acids, linked via amide or 'peptide' bonds;
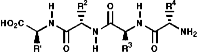
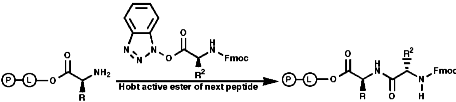
Peptide Synthesis
GeneralGenerally the process involves many steps for each peptide bond formed;
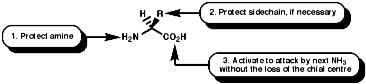
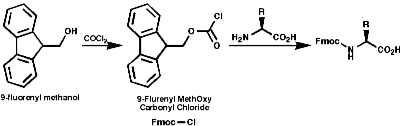
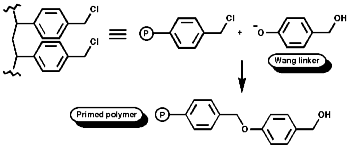
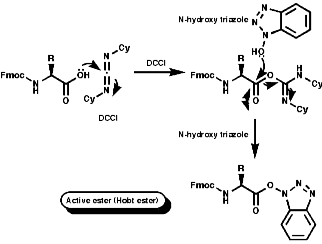
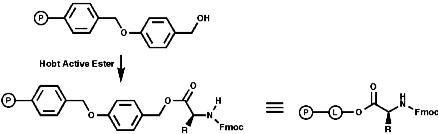
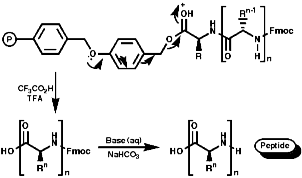
Merrifield Solid Phase Synthesis
The general principles of this method can be expressed as;
- Protection of NH2 group on amino acid;
- Resin, chloromethalated polystyrene, cross linked with 1% 1,4-divinyl benzene;
- Activation of the amino acid;
- Coupling of resin and amino acid;
- Deprotection of the amine;
- Coupling of resin/amino acid with another amino acid. Peptide growth;
- Cleavage of peptide from resin;
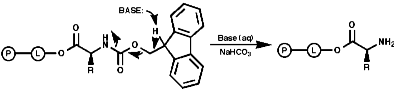
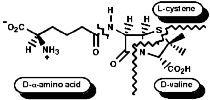
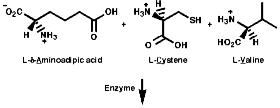
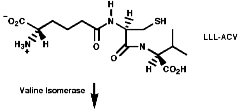
The biosynthesis of penicillin N, discussed earlier, is synthesised from its three constituent compounds via enzymic processes;
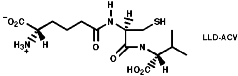
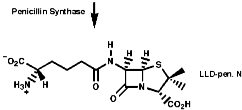
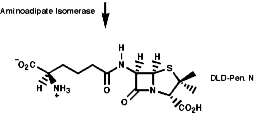
Summary
- Monomer for peptides and proteins
- N-protection / CO2H activation for peptide synthesis
- Streaker synthesis, RCHO / HCN / NH3 + hydrolysis
- Gabriel synthesis, actamidomalonate synthesis
- Monsanto syntheses, chiral hydrogenation
- Resin based solid phase peptide synthesis, Merrifield
Nitrogenous Natural Products and Alkaloid Biosynthesis
Introduction
Structural types of nitrogenous natural products are extremely varied and one of the most fascinating aspects of organic chemistry. Those commonly met with are;- Amino Acids, Peptides, Proteins and Derived Substances
- Vitamins
- Porphyrins, Chlorins and Cobalamins
These include structural proteins, enzymes, glycoproteins, and the peptide derived materials such as the b lactam antibiotics, the penicillins and cephalosporin C. For further information see Vollhardt chapter 26.
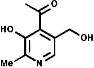
A miscellaneous collection of structures which are essential dietary constituents in mammals because of their inability to synthesise them. They have in common the fact they are usually cofactors for essential metabolic enzymes, e.g vitamin B6, the cofactor in the transamination process which introduces the amino-function into amino acids. For further information see Vollhardt chapter 25.
These are the tetrapyrrolic macrocyclic ligands for certain essential metals. They include; Haem, Fe2+ or 3+, for oxygen transport;
The coenzyme B12, Co2+ or 3+, for certain liver functions in animals;
Chlorophyll, Mg2+, for photosynthesis in plants;
For further information see Vollhardt chapter 25.
- Purines, Pyrimidines, Nucleosides, Nucleotides
- Alkaloids
The bases of the nucleic acids, DNA and RNA. These are covered in some detail in the biochemistry course in the second year. For further information see Vollhardt chapter 25.
Nitrogen containing natural compounds, other than those listed above, usually plant derived and frequently physiologically active. These are the classical folk medicines, some of which, e.g reserpine, a natural anti hypertensive agent and vinblastine, used in cancer treatment, are still important in medicine today. They are usually made in nature from amino acids precursors and are of many structural types. For further information see Vollhardt chapter 25.
Alkaloid Biosynthesis
Most alkaloids are plant products derived from amino acids. A widespread pathway is that based on the conversion of phenylalanine, tyrosine and DOPA to a variety of aromatic alkaloids including the opium alkaloids codeine and morphine. The sequence is based on the coupling of 2 phenylalanine, tyrosine or DOPA units to form 1-benzyl-1,2,3,4-tetrahydro-isoquinolines by a series of simple nitrogen based transformations of types familiar from the course. The basic reaction set, the ready interconversion in vivo of carbonyl groups, imines, enamines, and their derivatives, is;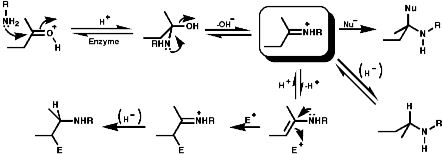
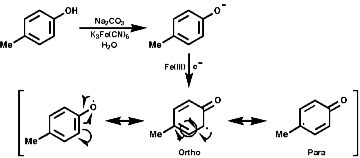
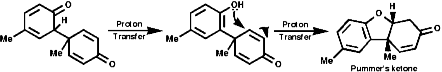
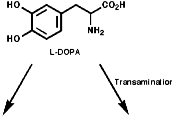
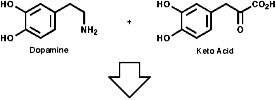
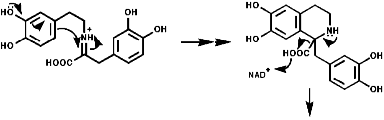
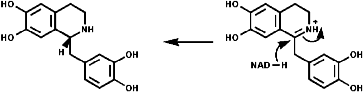
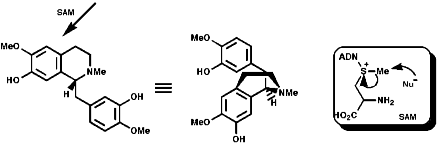
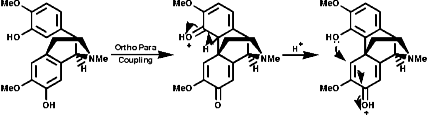
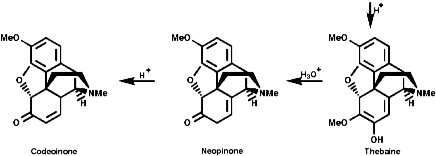
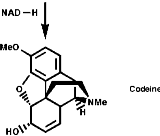
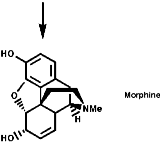
The sections on biosynthesis are intended to link with 'real' organic chemistry and to link with the polyfunctional and mechanistic chemistry presented in second year. They are for interest only and not for examination. purposes.
No comments:
Post a Comment